Zprávu, že umělá inteligence AlphaFold umí s vysokou přesností určovat prostorovou strukturu proteinů a tím způsobila revoluci v biologii, jste asi zaznamenali, plnila česká i světová média. Co přesně to znamená? Jak se umělá inteligence učí? Přišli strukturní biologové o práci? Na tyto a mnoho dalších otázek odpovídali odborníci z Univerzity Karlovy v panelové diskuzi s příznačným názvem Jáchyme, hoď ho do stroje.
Ilustrační prostorová struktura proteinu MAPK14 (Mitogen-activated protein kinase 14). Zdroj: Shutterstock
| Umělou inteligenci AlphaFold vyvíjí výzkumná společnost DeepMind, která spadá pod Google. Cílem DeepMind je pomocí umělé inteligence akcelerovat vědecký výzkum a tím i zlepšit chápání světa okolo nás. AlphaFold se zabývá podstatným a zároveň praktickým problémem – určováním prostorové struktury proteinů –, jehož vyřešení může pomoct biologii a medicíně. Podobný průlom se společnosti DeepMind podařil již v roce 2016, kdy umělá inteligence AlphaGo porazila ve hře go nejlepšího hráče světa – o desetiletí dříve, než se předpokládalo. |
„Myslím, že pro nás v oboru to svým významem bude něco jako přistání na Měsíci,“ přirovnával revoluční úspěch umělé inteligence AlphaFold hned v úvodu biolog Jan Černý z Přírodovědecké fakulty UK, který diskuzním večerem provázel. O tom, že se jedná opravdu o průlomové téma, svědčí i počet sledujících – online přenos na Facebooku a Zoomu ve čtvrtek v podvečer sledovala tisícovka zájemců.
Důležitost prostorové struktury proteinů
Určování prostorové struktury proteinů je i dnes jeden z nejtěžších úkolů biologie. Přitom prostorová struktura proteinů je zásadní – i jediná mutace (chyba) v primární sekvenci může narušit správné složení a tím i funkci proteinu. U AlphaFoldu se zmiňuje, že vyřešil padesát let starý biologický problém: „V roce 1972 získal C. B. Anfinsen Nobelovu cenu za chemii za popis vztahu sekvence a struktury proteinu. Anfinsenovo dogma říká, že strukturu proteinu je možné určit jen ze znalosti sekvence aminokyselin, v praxi je to ale stále velmi obtížné,“ řekl v úvodu své přednášky bioinformatik Marian Novotný z Přírodovědecké fakulty UK. Primární strukturu proteinů, pořadí aminokyselin, dnes již umíme velmi dobře a jednoduše určovat sekvenováním – sekvenci známe asi u 200 milionů proteinů. „Určování struktury je ale stále složité. Máme techniky, jako například rentgenovou krystalografii, NMR spektroskopii nebo kryoelektronovou mikroskopii, kterými prostorovou strukturu proteinu umíme experimentálně určit, je to ale velmi nákladné a často to trvá roky,“ vysvětloval Novotný, proč strukturu známe jen u 170 tisíc proteinů – pouhé tisíciny známých proteinových sekvencí.
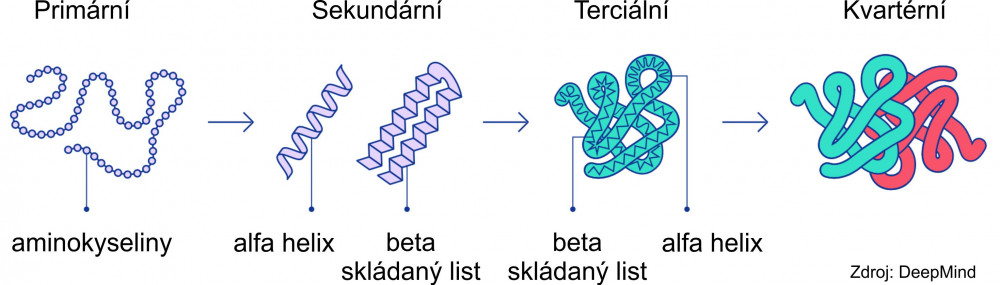
| Proteiny (bílkoviny) jsou základem všech živých organismů, kde plní řadu funkcí: stavební (například kolagen), transportní (přenašeč kyslíku hemoglobin), zajišťující pohyb (aktin a myosin), řídící a regulační (hormony) nebo ochranné (imunoglobuliny). Primární struktura proteinů je určena pořadím (sekvencí) 21 aminokyselin, které si můžeme představit jako korálky na šňůrce. Jednotlivé aminokyseliny spolu lokálně interagují a „šňůrka korálků se kroutí do preferovaných uspořádání“ – alfa helixů nebo beta skládaných listů spojených smyčkami, které tvoří sekundární strukturu. Uspořádání celého proteinového řetězce „celé šňůrky korálků“ v prostoru udává terciální struktura. Proteiny živých organismů často tvoří velké komplexy – kvartérní strukturu, kdy spolu funkčně interaguje několik podjednotek proteinů – „několik šňůrek korálků“. |
“Když chcete, aby se lidé v nějakém oboru zlepšili, vymyslete pro ně transparentní soutěž,“ komentoval s nadsázkou bioinformatik Novotný. Ale přesně to se v oboru strukturní biologie děje. Již od roku 1994 se každé dva roky koná unikátní vědecká soutěž CASP (Critical Assessment of Techniques for Protein Structure Prediction), ve které AlphaFold již podruhé zvítězil. „Je to taková olympiáda pro strukturní biology, která má zásadní vliv na rozvoj celého oboru,“ přirovnával Novotný.
Strukturní olympiáda pro umělou inteligenci
Organizátoři CASPu každé dva roky vyzvou strukturní biology, kteří jsou těsně před dokončením určení struktur proteinů tradičními metodami, aby jim poslali sekvence proteinů. Z těchto sekvencí se soutěžní týmy pomocí nejrůznějších výpočetních metod, v posledních letech především pomocí umělé inteligence a strojového učení, snaží predikovat strukturu proteinů. Ve výsledku se pak porovnají předpovědi umělé inteligence a experimentálně získané struktury daných proteinů, které vědci mezi tím dokončili, ale ještě nikde nepublikovali. Výsledky předpovědí umělé inteligence se hodnotí na škále od 0 do 100, kdy výsledky predikcí nad 95 bodů jsou srovnatelné s experimentálně určenými strukturami proteinů. Jednotlivé ročníky soutěže CASP se mezi sebou nemohou přímo porovnávat, protože každý ročník je jinak obtížný, ale v roce 2016 nejlepší týmy dosahovaly skóre 40, v roce 2018 se poprvé zúčastnil tým AlphaFold a dosáhl skóre kolem 70 bodů, letos v CASP 14 nasazený AlphaFold 2 dosáhl průměrného skóre dokonce 92,4 bodů – mnohem více než ostatní soutěžní týmy.
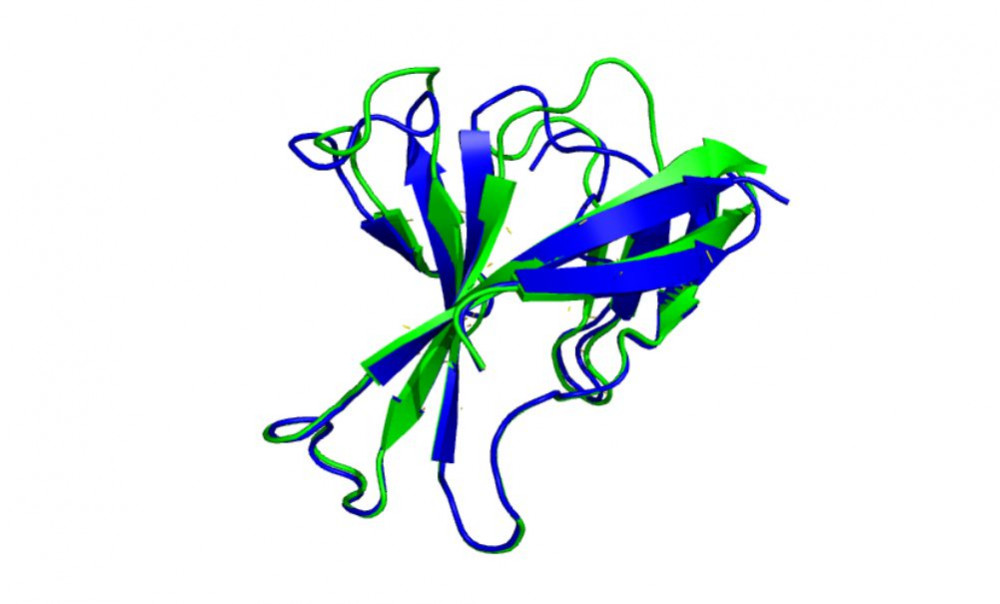
Srovnání predikce AlphaFold2 (zeleně) a experimentálně určené struktury (modře) u proteinu orf8 viru SARS-Cov-2. Autor: Marian Novotný.
Jak se umělá inteligence učí
„Strojové učení je honosný název pro učení z dat. Je to podobné jako když se například malé dítě učí rozlišovat mezi kočkou a psem – mnohokrát mu ukážete různé příklady, co je pes a co kočka, a dítě se postupně naučí, jak je rozlišit,“ přirovnával odborník na vývoj bioinformatických algoritmů a nástrojů David Hoksza z Matematicko-fyzikální fakulty UK, jak se AlphaFold učil ze sekvencí známých proteinových struktur. „Hluboké neuronové sítě si můžeme představit jako sít jednotlivých neuronů – výpočetních jednotek, které ze vstupních dat určitým procesem, který může mít mnoho kroků-vrstev, vytvoří výstup,“ popisuje.
Předchozí verze AlphaFoldu uvedená v CASPu13 pro své učení využívala proteinové sekvence, které jsou volně přístupné v databázích a jako první se učila ze vzdáleností mezi atomy. „AlphaFold 1 předpovídá vzdálenost a torzní úhly mezi každou dvojicí aminokyselin ve výsledné struktuře proteinu, přičemž počítá se všemi aminokyselinami najednou, protože se vzájemně ovlivňují. Na proteiny se tak dívají komplexně, což je blízké biologii, ale extrémně náročné na výpočet, v tom umělá inteligence významně pomáhá,,“ vysvětloval Hoksza.
Za dva roky došlo k výraznému zlepšení nejen v týmu AlphaFold, ale i u dalších týmů. „Na CASP panuje soutěživě spolupracující prostředí, pro celý obor je důležité, že se týmy pravidelně potkávají. Například před dvěma lety AlphaFold pro hluboké učení jako první použil vzdálenost mezi atomy, v letošním ročníku soutěže to již používalo mnoho dalších týmů,“ komentoval Novotný
„O letos uvedeném AlphaFold 2 toho zatím víme velmi málo, byl představen bez detailů, ale předpokládám, že bude fungovat asi výrazně jinak než ostatní dosud známé přístupy,“ shrnul Hoksza. „AlphaFold 2 například zcela unikátně umí určit, zda je daná predikce dobrá nebo ne – jak moc si je jistý s predikcí i na úrovni jednotlivých aminokyselin.“
AlphaFold pomůže, ale strukturního biologa nenahradí
„Umělá inteligence může zásadním způsobem pomoci při řešení proteinových struktur a otevřít tak i mnoho zcela nových možností, ale strukturní biologové o práci rozhodně nepřišli,“ komentoval strukturní biolog Václav Veverka z Ústavu organické chemie a biochemie Akademie věd ČR a Přírodovědecké fakulty UK. „Navíc bez struktur vyřešených experimentálními technikami by žádná umělá inteligence nebyla, neměla by se z čeho učit. Dobře je to vidět na výsledcích – AlphaFold 2 velmi dobře řeší jednodoménové proteiny, protože těch je i hodně v databázích, ze kterých se učil,“ dodal Veverka. Sám vidí největší pomoc umělé inteligence při experimentálním určování struktur pomocí hmotnostní spektrometrie, kryoelektronové mikroskopie či rentgenové krystalografie. „Naopak dynamické proteiny budou pro umělou inteligenci vždy problém – NMR bude mít ještě dlouho své nezastupitelné místo,“ uvedl strukturní biolog.
AlphaFold si také (zatím) neumí poradit s velkými multiproteinovými komplexy nebo naopak s malými detaily na úrovni jednotlivých aminokyselin. „Pravě tyto detaily jsou důležité při hledání nových léčiv nebo určování vlivu jednotlivých mutací, očekávám ale, že se tyto detailní predikce budou zlepšovat,“ shrnul Veverka. AlphaFold také neříká nic o samotném skládání proteinů. „Je to taková černá skříňka – ze vstupní sekvence vám určí strukturu, ale vůbec nevíme jak,“ doplnil Novotný.
Co můžeme očekávat za dva roky? „V předchozích ročnících CASP soutěžní týmy vždy přišly s něčím novým – novou technologií nebo aplikací nové architektury neuronových sítí. Tipuji, že do dvou let přijde nová architektura neuronových sítích, která ještě vylepší predikce,“ sdílel svůj pohled David Hoksza. Všichni přednášející, se shodli, že umělá inteligence může zásadním způsobem pomoci při řešení proteinových struktur a může otevřít zcela nové možnosti ať už v oblasti základního výzkumu, ale i vývoje nových léčiv či léčby onemocnění. „Úspěch AlphaFoldu můžeme svým významem přirovnat například k osekvenování lidského genomu, může to způsobit revoluci v biologii, ale přesný rozsah nebo podobu si zatím nikdo neumí představit,“ shrnul bioinformatik Novotný.















